Understanding gene regulation at the transcriptional level is critical to understanding complex biological systems and human disease. In virtually all organisms gene regulation is mediated by a “regulatory code” in which distinct combinations of specific transcription factors (TFs) collaborate to regulate the expression of individual genes. This code is complex and not readily obvious from sequences alone. It likely involves many cis-regulatory modules (CRMs) that exist both upstream and within genes. Data from the ENCODE and modENCODE projects suggests that the amount of cis-regulatory sequence may exceed that of the genes themselves. In addition, mounting evidence suggests that major differences between individuals and species lies at the level of gene regulation and that changes in cis-regulatory sequences are responsible for these effects. As such, it is important to map and understand how sequence variations in individuals are responsible for mediating differences in gene expression and their phenotypic consequences. The goal of my research is to understand the biological mechanisms underlying transcriptional regulation and how human variation at regulatory regions affects this process.
OUR PROJECTS
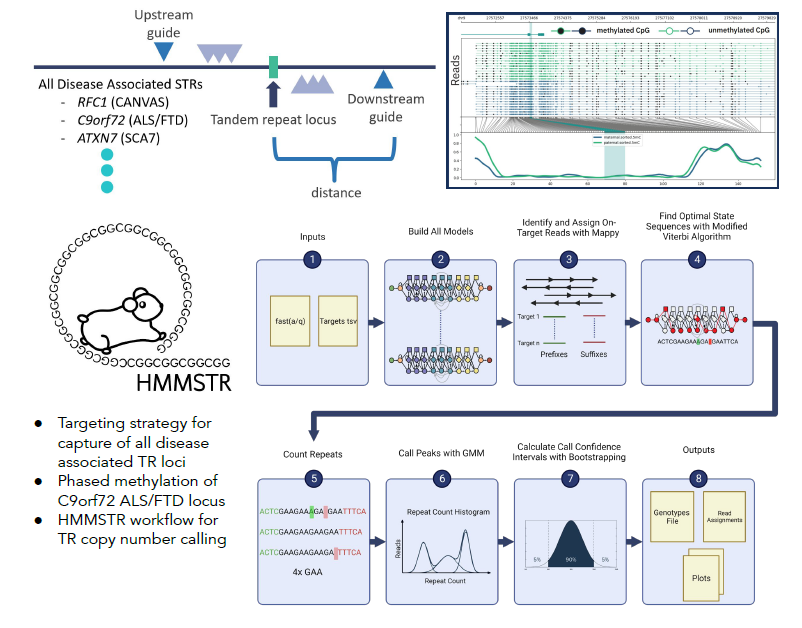
Targeted characterization of tandem repeats
Known tandem repeats (TRs) makeup 3% of the human genome and are highly variable across individuals. Tandem repeats are intrinsically unstable, and their expansion is known to cause over 50 human diseases including ALS, Ataxia, and Huntington’s Disease. While combined these disorders have a high prevalence, characterization and discovery of these loci have proven elusive with short read sequencing techniques due to their repetitive nature. Long read sequencing technologies such as Oxford Nanopore Technologies produce reads up to 2Mb allowing for sequencing repeat elements in their entirety, however they have relatively high error rates which poses another obstacle to accurate quantification of TR copy number. We aim to more accurately characterize tandem repeat regions, both at healthy and pathogenic lengths, with a combination of targeted Nanopore sequencing and improved computational methods.
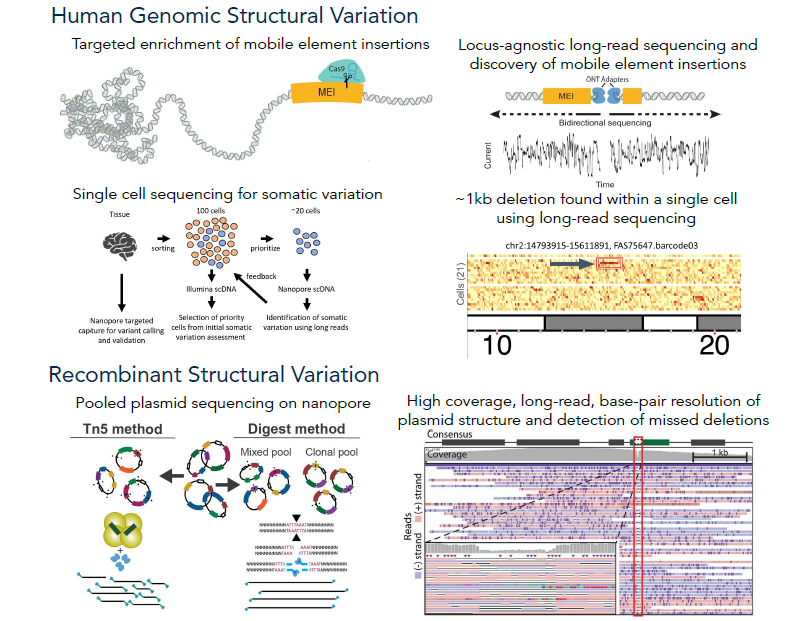
Nanopore sequencing for variant characterization
It is known that the human genome varies not only from person to person, but also within individuals. This variation is termed somatic mosaicism as it occurs after conception and leads to differing amounts of variation in cells and tissues. High-frequency variation has been linked to diseases like cancer, but each type of human tissue shows varied rates of this mosaicism. We are working to systematically investigate somatic variation across tissues via the SMaHT initiative, as well as improving and optimizing existing bioinformatic and DNA sequencing methods. This will likely lead to the discovery of previously overlooked classes of somatic variation. It has been found that somatic mutations and mobile elements insertions (MEIs) from retrotransposons in the brains of people with Alzheimer's disease could be a possible cause of this neurodegenerative disease. These mutations and MEIs are most commonly found in neurons within the cerebral cortex, which is involved in the progression of Alzheimer's. However, it's challenging to measure MEIs because of their repetitive nature, and the sequencing technology needed to map them is inadequate and/or expensive. Now, we're using long-read sequencing to improve MEI identification, specifically L1Hs, Alus and SVAs, and pinpoint where these elements are found in the genome by studying the brains of Alzheimer's disease patients.
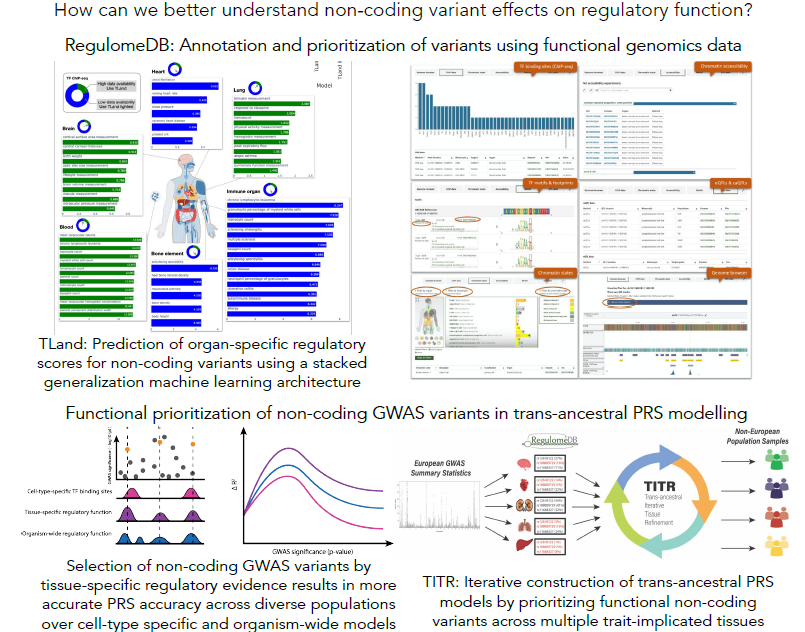
The impact of genetic variation on gene regulation
Genome-wide association studies (GWAS) have identified numerous genetic variants associated with complex disease and traits. However, understanding the functional consequences of disease-associated variations is a significant challenge in human genetics. This is particularly true for variants in non-coding genomic regions, which accounts for over 90% of all GWAS variants. High-throughput functional genomic assays like ChIP-seq and DNase-seq can help characterize regulatory elements interspersed throughout the non-coding genome. We have developed a number of machine learning tools - SURF, TURF, and TLand - that combine evidence from multiple functional genomic assays from the ENCODE project to predict regulatory function of non-coding variants for both general functional activity and cell- and tissue-specific activity. These data are summarized on RegulomeDB.org to aid researchers in studying regulatory variants. We also leverage these functional predictions to construct genetic risk models that highlight variants with high functional probabilities to assess disease liability in understudied non-European populations. The hypothesis is regulatory programs are common between diverse ancestries and that by prioritizing high confidence functional variants in genetic risk modeling, we can better circumvent common issues like differences in linkage patterns in trans-ancestral risk modeling.
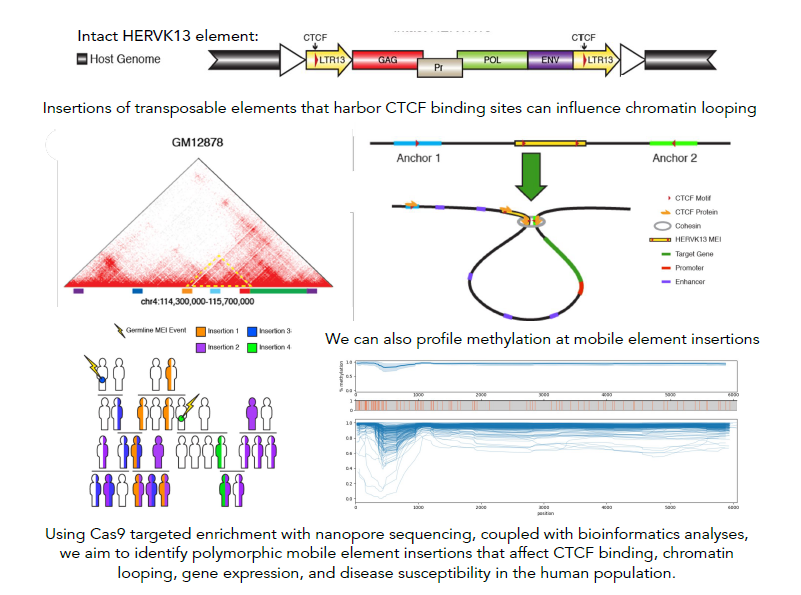
Mobile element derived chromatin looping variability in the human population
Our research centers on the exploration of insertion polymorphisms of transposable elements (TEs) - DNA sequences that are or were capable of multiplying and/or changing their position in the genome. These TEs, constituting at least 45% of the human genome, have been traditionally mislabeled as 'junk DNA' with no proposed influence on human traits. However, a growing number of studies indicate that certain TEs could not only be associated with disease susceptibility and progression but could also impact critical regulatory functions within a healthy cell. Previous research conducted in our lab has revealed that multiple TEs within the human genome feature binding motifs for CTCF, the protein that plays a vital role in defining the 3D structure of mammalian genomes by facilitating the loop formation between distal genomic sequences through the cohesion complex. Three-dimensional chromatin structure has been linked to many critical genomic functions, including the regulation of gene expression, which can have a major impact on phenotype, both in health and disease. We posit that the variation in TE insertion at the population level might be a significant, yet underexplored, factor influencing CTCF binding and chromatin looping among humans. Using bioinformatics approaches and computational tools as well as innovative methods for sequencing specific types of TE insertions via genome-wide capture using short guide RNAs (sgRNAs), followed by long-read sequencing with Oxford Nanopore sequencing technology, we aim to gain further insights into the nature and extent of differences in CTCF binding and chromatin 3D structure that TE activity introduces at a population level.
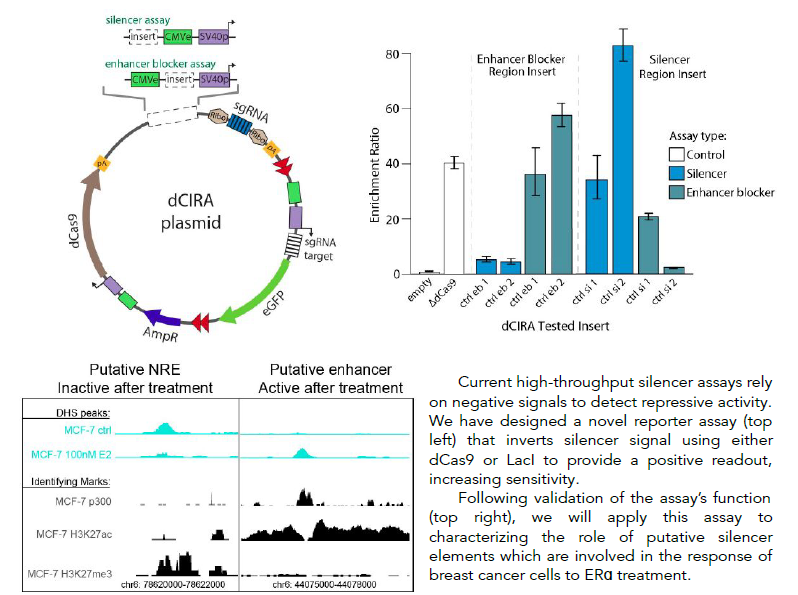
High-throughput inverted reporter assay for characterization of silencers and enhancer blockers
Cis-regulatory elements (CREs) are short stretches of DNA sequence located outside of genes, which help control gene expression. They are essential to controlling the proper timing, location, and order of gene expression, which determines cell identity and function. CREs include four known types of element: promoters, enhancers, silencers and enhancer blockers. While some studies estimate that there are tens of thousands of silencers and enhancer blockers distributed throughout the human genome, very few of these elements have been mapped until recently and their function has been characterized in only a few common cell lines. Previous work on cis-regulatory element function has focused primarily on the role of enhancer activity and its disruption, while the roles of negative regulatory elements such as silencers and enhancer blockers are less well understood. A major reason for this disparity is the lack of suitable massively parallel reporter assays (MPRA) designed for silencer testing. Existing assays suffer from inherent design limitations, leading to high false positive or false negative rates, or prohibitive sequencing requirements. The Boyle lab is developing a novel pair of innovative high-throughput reporter assays designed specifically for functional testing of silencers and enhancer blockers, that improve on the limitations of existing methods. They are designed for increased sensitivity, specificity, and decreased cell number and sequencing requirements through the use of a novel dCas9- or LacI-based repression signal inversion approach. Development of these assays will facilitate mapping and characterization of this important class of regulatory elements.
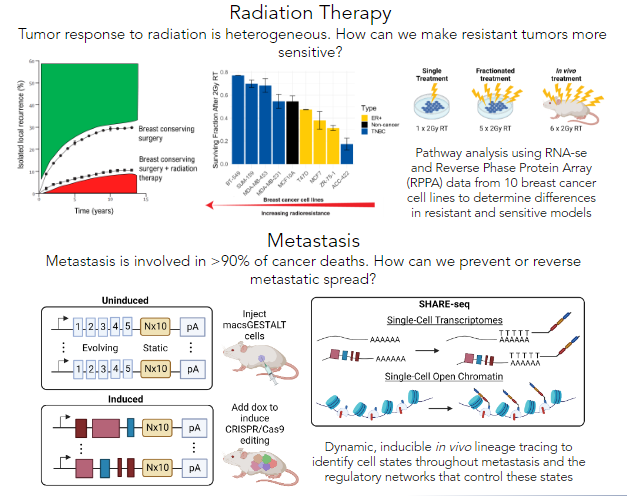
Improving breast cancer patient prognosis with targeted therapies
Breast cancer is the most diagnosed cancer in the world, and remains the most deadly for women. Clinical management of breast cancer includes radiation therapy as a mainstay, with upwards of 85% of women receiving radiation therapy as part of their treatment regimen after breast conserving surgery. Although effective, over 10% of women will develop a local recurrence despite radiation therapy. Unfortunately, the molecular mechanisms that underly radiation response and intrinsic radioresistance or radiosensitivity are poorly understood. We are leveraging multi-omics data to interrogate these mechanisms.
Additionally, metastasis is involved in over 90% of cancer-related deaths. Triple-negative breast cancer is known for earlier disease onset and a higher propensity to metastasize relative to other breast cancer subtypes, making it more likely that it will metastasize before it can be diagnosed. Thus, knowing the phenotypic states of cells that can successfully metastasize and the transcriptional regulatory networks that govern them is invaluable knowledge to prevent and reverse metastasis in individuals with this disease, ultimately improving patient prognosis.